Study reveals potential therapeutic target for genetic liver disease

Northwestern Medicine investigators have discovered the molecular mechanisms underlying a rare genetic mutation that increases the risk of severe liver disease, according to findings published in Molecular Cell.
The study, led by Deyu Fang, Ph.D., the Hosmer Allen Johnson Professor of Pathology, may reveal potential therapeutic targets for treating the disease.
Alpha-1 antitrypsin (AAT) deficiency is a rare genetic mutation that can increase the risk of developing several diseases, including severe liver disease. One in 3,500 people in the U.S. are diagnosed with the genetic disorder annually.
The AAT protein is made by the liver and moves into bloodstream to protect the liver and other organs from infection. AAT deficiency, however, can cause low levels of AAT in the bloodstream and a build-up of the protein in the liver, which can be life-threatening.
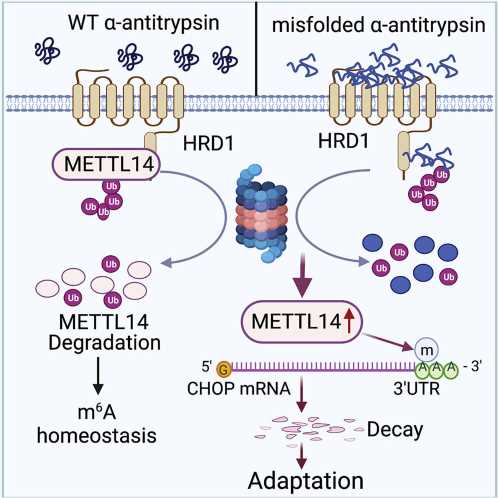
AAT deficiency is caused by a mutation in the serine protease inhibitor 1A (SERPINA1) gene, which produces AAT. On a molecular scale, the mutation results in toxic unfolded proteins that accumulate within the endoplasmic reticulum of hepatocytes—specialized liver cells—and triggers an unfolded protein response for cell stress adaption.
The unfolded protein response is essential to restore the cell’s normal function by stopping protein translation, to degrade misfolded proteins and to activate beneficial cell signaling pathways. Failure of the unfolded protein response, however, induces apoptosis, or cell death, as well as damage to surrounding tissue or organs—in this case, the liver.
The molecular mechanisms that determine how this unfolded protein response selects stress adaption over apoptosis have remained unknown.
In the current study, Fang and colleagues used immunofluorescence to study liver cell lines from mouse models of AAT-induced liver disease.
They found that the accumulation of unfolded proteins upregulated the expression of the metabolic enzyme N6-adenosine-methyltransferase-14, or METTL14, in the mouse models. METTL14 also suppressed C/EBP-homologous (CHOP) protein-induced apoptosis for endoplasmic reticulum stress adaption.
“Mice with liver specific METTL14 deletion are highly susceptible to both acute pharmacological and AAT deficiency-induced endoplasmic reticulum proteotoxic stress and liver injury,” Fang said.
Source: Read Full Article


