
Multiomics identifies genomic risk factors for COVID-19
As of June 15, 2022, over 540 million people have been infected with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus responsible for the coronavirus disease 2019 (COVID-19), of which more than 6.3 million have died.
Age, as well as the presence of certain comorbidities, have been associated with an increased risk of severe COVID-19 and death. Various other factors, including specific genetic predispositions, are also thought to increase an individual’s likelihood of severe COVID-19; however, this association remains poorly understood.
 Study: Multiomic analysis reveals cell-type-specific molecular determinants of COVID-19 severity. Image Credit: Alpha Tauri 3D Graphics / Shutterstock
Study: Multiomic analysis reveals cell-type-specific molecular determinants of COVID-19 severity. Image Credit: Alpha Tauri 3D Graphics / Shutterstock
Background
Previous observational studies have found that COVID-19 severity can be correlated to heightened levels of several immune cells, including CD8 T-cells, CD19 B-cells, eosinophils, myeloid cells, as well as several different subtypes of immune cells, such as adaptive natural killer (NK) cells. Similar findings have been confirmed with transcriptomic studies; however, these alterations in immune cell types were likely due to the acute infection and do not necessarily represent a predictive marker for severe disease.
The hyperinflammatory response that occurs within the lungs of patients experiencing severe COVID-19 is a primary cause of morbidity and mortality in these patients. As a result, most of the interventions that are currently approved for use in treating severe COVID-19 are repurposed drugs that aim to suppress this immune response.
Since SARS-CoV-2 has become an endemic virus, it is essential for researchers to better understand the precise mechanisms involved in the pathogenesis of COVID-19, particularly in severe cases. This information would subsequently contribute to the development of more potent and effective therapeutics.
About the study
In a recent Cell Systems study, researchers utilized the RefMap machine learning algorithm to correlate known genomic variations for severe COVID-19 obtained from large-scale genome-wide association studies (GWAS) with single-cell multiome profiling of human lungs. One of the key advantages of this combined approach is that it allowed the researchers to identify functional regions within the genome associated with severe COVID-19 and integrate this information with cell-type-specific functions.
Furthermore, RefMap reduces the size of the search space by modeling the entire genetic architecture of critical diseases through the use of a unified probabilistic model. Thus, by capturing more complex genetic structures, researchers can avoid a multiple testing correction, which ultimately enhances its statistical power.
For the human lung samples, both single nucleus ribonucleic acid sequencing (snRNA-seq) and single nucleus assay for transpose-accessible chromatin using sequencing (snATAC-seq) were performed.
A total of 19 different cell types were identified through these sequencing assays, of which included various epithelial, endothelial, and hematopoietic cell types. RefMap was then used to correlate disease-associated genomic regions from the COVID-19 GWAS data with reported peaks of these 19 cell types.
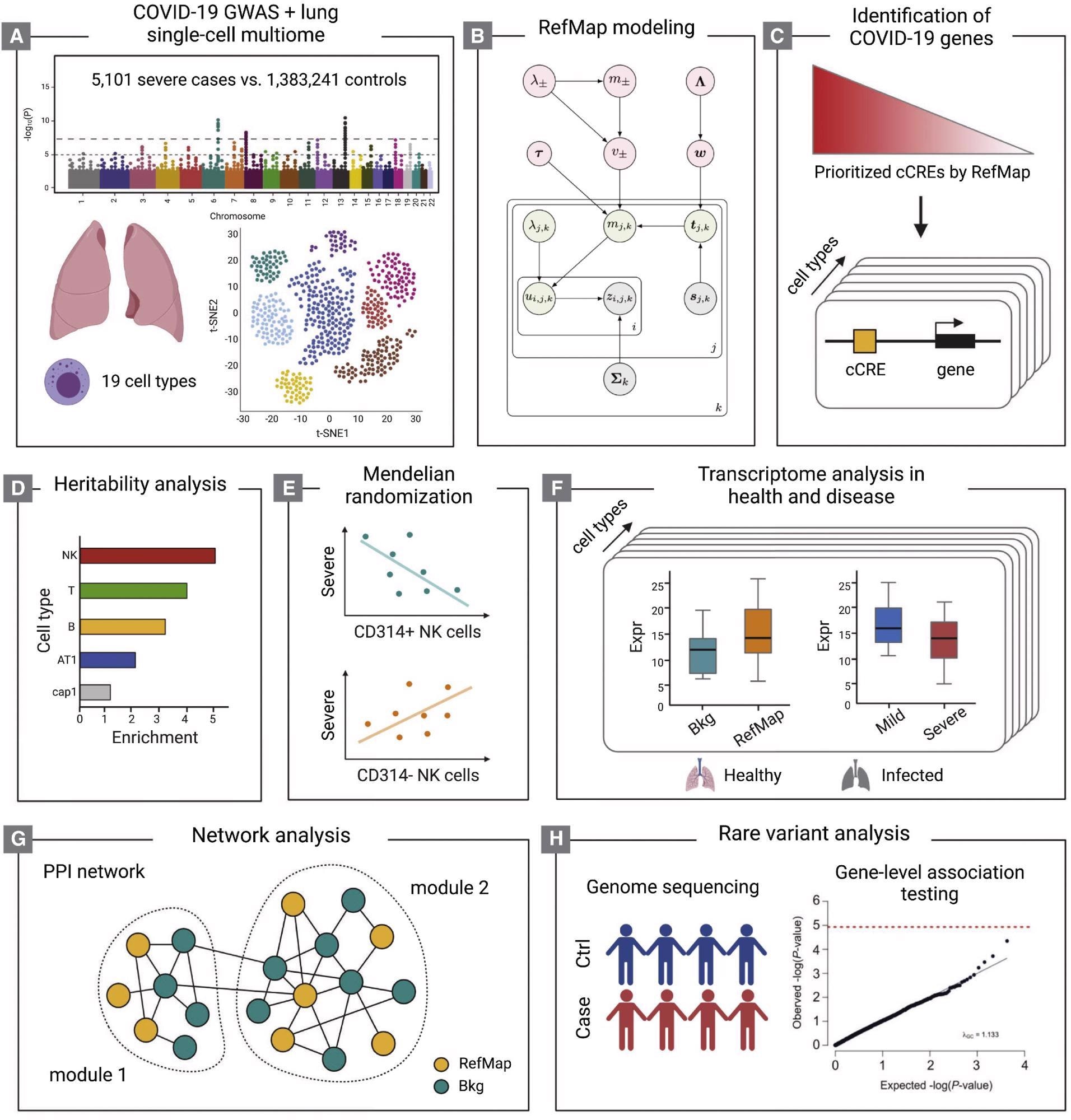 Schematic of the study design (A-H) The COVID-19 GWAS and human lung single-cell multiome (A) are integrated by the RefMap model shown in (B), where gray nodes represent observations, green nodes are local hidden variables, and pink nodes indicate global hidden variables (STAR Methods). Cell-type-specific risk genes are mapped using single-cell multiome profiling (C). Heritability analysis (D), Mendelian randomization (E), transcriptome analysis (F), and network analysis (G) together characterize the functional importance of RefMap genes, particularly for NK cells, in severe COVID-19. Rare variant analysis (H) orthogonally supports the role of NK cells in severe disease. cCRE, candidate cis-regulatory element.
Schematic of the study design (A-H) The COVID-19 GWAS and human lung single-cell multiome (A) are integrated by the RefMap model shown in (B), where gray nodes represent observations, green nodes are local hidden variables, and pink nodes indicate global hidden variables (STAR Methods). Cell-type-specific risk genes are mapped using single-cell multiome profiling (C). Heritability analysis (D), Mendelian randomization (E), transcriptome analysis (F), and network analysis (G) together characterize the functional importance of RefMap genes, particularly for NK cells, in severe COVID-19. Rare variant analysis (H) orthogonally supports the role of NK cells in severe disease. cCRE, candidate cis-regulatory element.
Study findings
Taken together, the researchers identified 1,370 genes that were associated with severe COVID-19, with hematopoietic cells containing the largest number of unique RefMap regions and genes of all cell types. These genes accounted for 77% of the single nucleotide polymorphism (SNP)-based heritability for severe COVID-19. Some of the key RefMap COVID-19 genes that were associated with severe disease included increased expression of LZTFL1 in ciliated epithelial cells and increased activity of the metalloprotease ADAMP9.
The researchers were then interested in characterizing the functional roles of these severe COVID-19 genes. To this end, four transcriptional factors (TFs) including CUX1, TCF12, ZEB1, and ZEB2 binding motifs were found to be enriched in at least one of the 19 cell types.
ZEB2, in particular, was only enriched in NK cell risk regions. Since ZEB2 plays an important role in the maturation of NK cells, the authors concluded that severe COVID-19 may arise due to the failed maturation of NK cells.
Further analysis into the expression of NK cell subtypes in the severe COVID-19 genes led to the identification of an increased expression of CD56bright NK cells, particularly when compared with the expression of CD56dim cells. Whereas CD56bright NK cells are responsible for the production of cytokines and modulating the immune response, CD56dim NK cells are directly cytotoxic.
As compared to the total set of RefMap NK-cell genes, CD56bright NK cells genes were found to be highly enriched with heritability for severe COVID-19 to a greater extent than that of any other profiled cell type. This finding indicates that altered functioning and/or deficiency of CD56bright NK cells prevents the efficient production of important cytokines, particularly interferon γ (IFN- γ).
Conclusions
The genetics-based approach utilized in the current study provided evidence that correlates with previous observational reports on the relationship between altered CD56bright NK cell function and severe COVID-19. Due to the important role of CD56bright NK cells in the innate immune response, their deficiency may precipitate the uncontrolled viral replication of SARS-CoV-2.
- Zhang, S., Cooper-Knock, J., Weimer, A. K., et al. (2022). Multiomic analysis reveals cell-type-speicfic molecular determinants of COVID-19 severity. Cell Systems. doi:10.1016/j.cels.2022.05.007, https://www.sciencedirect.com/science/article/pii/S2405471222002289
Posted in: Device / Technology News | Medical Research News | Disease/Infection News
Tags: Assay, Cell, Chromatin, Coronavirus, Coronavirus Disease COVID-19, Cytokines, Drugs, Genes, Genetic, Genetics, Genome, Genomic, Immune Response, Interferon, Lungs, Machine Learning, Mortality, Multiomics, Nucleotide, Respiratory, Ribonucleic Acid, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Single Nucleotide Polymorphism, Syndrome, Therapeutics, Virus

Written by
Benedette Cuffari
After completing her Bachelor of Science in Toxicology with two minors in Spanish and Chemistry in 2016, Benedette continued her studies to complete her Master of Science in Toxicology in May of 2018.During graduate school, Benedette investigated the dermatotoxicity of mechlorethamine and bendamustine; two nitrogen mustard alkylating agents that are used in anticancer therapy.
Source: Read Full Article


